CPVS enables clients to develop regulatory strategies that focus on identifying and acquiring the scientific evidence needed to support the safety and efficacy of mixes.
We assist in discovering and addressing critical path issues that could potentially delay development timelines and we help to clarify the obstacles to registration. We provide strategic guidance in marketing authorisation applications (MAAs), clinical (efficacy and safety) summaries, overviews, and labelling development.
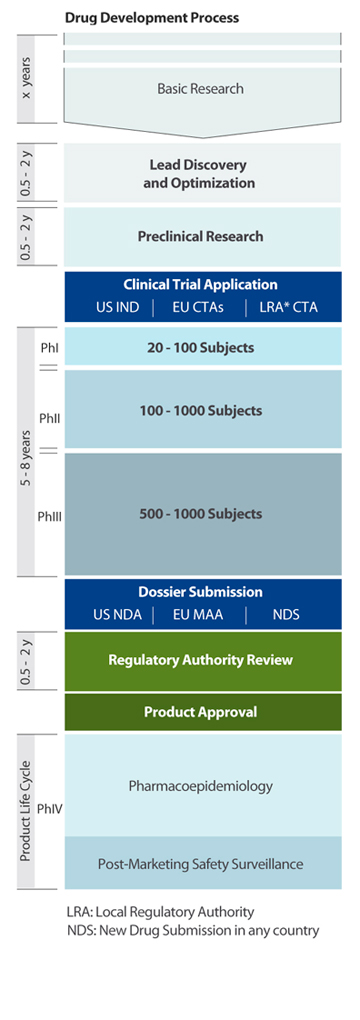 Drug Development Strategy & Regulatory Input in Clinical Programs
Drug Development Strategy & Regulatory Input in Clinical Programs
• Strategic development and execution of comprehensive drug development programs based on the compound’s Target Product Profile and the regulatory landscape.
• Strategic input in U.S. FDA meeting planning (pre-IND/IND, end-of-Phase I, II, III and pre-New Drug Application (NDA) meetings); briefing documents preparation and review.
• Guidance in the preparation for European scientific advice (including European Medicines Agency [EMA] and national procedures): briefing documents preparation and review.
• Clinical Trial Application (CTA) and U.S. IND preparation, review and filing.
• Guidance in contract research organisation (CRO) selection for individual clinical trials.
• Full clinical programs execution and oversight.
Creation of Labelling for Inclusion in Original U.S. NDA and MAA
• Development of initial U.S. Package Insert, European Summary of Product Characteristics (SPC), patient leaflet and other local product information documents based on data from product development, the most current labelling guidelines and regulations, product therapeutic areas, recent approvals, and regulatory trends.
Due Diligence
• Assistance in due diligence activities (European Union, U.S.) for products in all phases of development and commercialisation.
• Identification of business partners for critical business functions, such as drug development, distribution, and co-marketing.